
中国学科发展战略·半导体物理学进展
第一节 能带计算方法的沿革和现况
量子力学是 20 世纪物理学最重要的进展之一,它被广泛地应用到原子、分子、固态体系中,并取得了巨大的成功。尤其是自 20 世纪上半叶基于量子力学的半导体能带理论建立以来,它引领了半导体物理、材料和器件的发展,推动了微电子、光电子工业的进步,从而深刻地改变了人类的生活方式和历史进程。
典型的固态体系,如半导体材料,是由大量(1024)原子、分子组成的。对这样具有大量自由度的体系,采用量子力学的方法来精确求解电子和原子核的运动原则上是可行的,实际上是做不到的。物理学家们聪明地采用玻恩-奥本海默(Born-Oppenheimer)近似方法,把原子核的运动和电子分开来求解。即便如此,电子的能谱仍然无法计算。在朗道费米液体理论的框架下,电子的行为可以用无相互作用的准粒子图像来描述。因此,需要发展用以量子力学为基础的能带计算方法来了解和描述材料的原子结构及其复杂的物性,将电子与电子及电子与原子的多体相互作用在量子力学理论框架内进行综合处理。半导体能带理论和计算不仅能为材料的性能提供详细准确的信息,同时能够预测和挖掘新型功能材料。正是这种计算手段推动了近年来纳米功能材料、拓扑绝缘体和二维材料实验研究的进展。
从 20 世纪 80 年代开始,随着半导体制备技术的进步和提高,人们对半导体纳米结构的研究兴趣日益浓厚。当半导体材料缩小到纳米尺度时,量子效应开始显现,出现了许多有趣的新现象。半导体纳米结构中类原子的能级结构和光谱、声子瓶颈、能带计算和分子动力学模拟在半导体材料结构特性、光学和电学性质的研究中都发挥着十分重要的作用。纳米体系中含有较多的粒子(100~100 000),这对半导体能带理论和计算提出了挑战。以量子力学为基础的材料模拟的计算方法可分为第一性原理计算方法和半经验方法(其中包括 KP 理论、经验赝势和紧束缚等方法)。每种方法都有其自身的优势和弱点,如前者具有可靠性和预测性,但所研究的体系大小受到很大的限制,而半经验方法在研究电子低能激发模式、解析推导和分析、大尺度复杂纳米体系及动力学模拟计算上具有很大优势,但其可靠性及预测性则取决于近似程度及参数的优化。
半导体能带计算是半导体物理的理论和实验的基础。第一性原理计算方法是最强有力的工具之一。但是,由于在该方法中低估了交换关联作用,对能隙计算误差较大。自 20 世纪 80 年代以来,人们孜孜以求地改进该方法, 以获得较为准确的能隙及相关的光学性质,其中较为成功的方法是 GW 方法和 HSE 方法。
Hohenberg-Kohn(HK)定理证明,多体系统中每个电子上的定域外势V(r) 仅仅对应于一个基态密度 (r),多粒子体系基态能量可写成基态电子密度的泛函,并不需要全电子波函数的完备知识,从而将求解多体薛定谔方程的问题严格转换为使 HK 能量泛函关于电荷密度为最小值的变分问题 [1]。为便于实际计算,Kohn-Sham 引入无相互作用体系的有效势V KS(r)模拟真实体系的基态电荷密度,导出著名的 Kohn-Sham 自洽方程组 [2]:
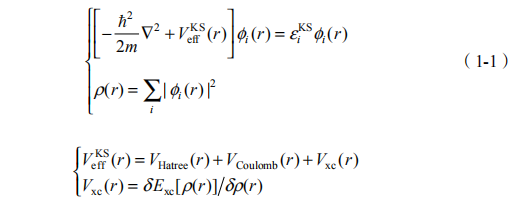
由于对上述方程组中交换关联能 Exc 的形式并不清楚,Kohn-Sham 提出以局域密度近似(LDA)计算 Exc ,即以均匀密度电子气局域描述非均匀密度电子气。由于非均匀密度电子气特征屏蔽长度与均匀电子气系统差别极小,且二者交换关联空穴满足的求和规则一致,密度泛函理论-局域密度近似(DFT-LDA)对于非均匀多体系统的基态性质的描述获得了超乎预期的成功。现在,人们已经可以对晶格结构、晶格常数、杨氏模量、价带结构以及晶格振动模式等基态性质进行精确的计算和预言。
但是,对于多粒子体系中的绝大多数可观测量,如光吸收谱、光反射谱、激子等激发态,DFT-LDA 近似原则上不能够正确地描述。其主要的表现在于,对于半导体的带隙,LDA 会严重低估 [3],而对于金属占据态的带宽,LDA 则会高估。究其原因,DFT-LDA 只是描述粒子数 N 不变的基态理论,而且其 Kohn-Sham 能量本征值 KS 没有严格的物理体系能量本征值的意义。使用 DFT-LDA 预言带隙时,Egks= cKS( N ) - v KS( N ),体系粒子数 N 不变,而实际物理中激发态是系统基态对外界微扰的响应,这种响应伴随有准粒子的产生和湮灭。最简单的形式是在占据态湮灭一个电子而会在未占据态产生一个电子。因此,对于占据态应考虑总能量差 E(N) - E(N-1),而对于未占据态应考虑总能量差 E(N+1) - E(N)。所以,真实的带隙应该为Eg= C (N +1) - V (N ) ,其与 Kohn-Sham 带隙的关系可以表达为
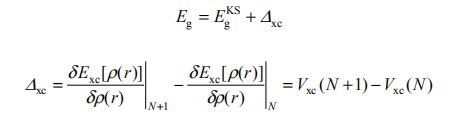
即 N+1 与 N 电子体系交换关联势的不连续差值。因此,DFT-LDA 总是系统地低估半导体的带隙 [4]。而因为 DFT-LDA 以无相互作用的均匀电子气有效势V KS(r)近似描述有相互作用非均匀多体系统,其对金属占据态带宽的描述更接近自由电子气的行为,从而总是系统地高估占据态带宽。关于这一现象, Northrup 和 Louie 曾在碱金属中有详细的阐述 [5]。
尽管如此,激发态问题毕竟是体系基态性质对于外界微扰的响应,处理激发态的理论也将与传统的静态密度泛函理论有天然的密切关联。以下我们将以密度泛函理论为基础,以格林函数为手段,研究激发态第一性原理计算方法的发展、沿革及所取得的成就。
第二节 MBGFT 方法与 GW 近似
依据场论的思想,密度泛函理论可视为一种平均场近似,而对于实际的有相互作用的多体系统,更为准确的语言是准粒子图像。由于正电荷背景的存在,电子与电子间的长程库仑相互作用受到屏蔽,长程库仑相互作用的多体系统变成了弱屏蔽库仑相互作用的准粒子系统。通过构建准粒子的类Kohn-Sham 方程,人们便可以描述包含较为准确的交换关联重要的激发态性质。因为采用量子场论中格林函数方法来描述,所以,这一方法又称为多体格林函数方法(MBGFT)[4,6,7]。
该方法于 1965 年由 Hedin 为解决电子气问题而提出 [8]。Hedin 提出了一组四个闭合的微积分方程,可与 Dyson 方程联立严格求解自能。这五个方程涉及的物理量包括:单粒子格林函数 G、自能 、屏蔽库仑相互作用 W、不可约极化传播矢 P 和顶角函数 。
单粒子格林函数 G(xt,x′t′ ) 作为准粒子的传播矢,描述的是准粒子由一时空点 (xt) 到另一时空点 (x′t′) 的产生、传播和湮灭的过程。其表达式为
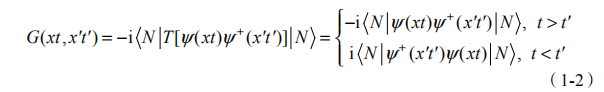
其中, x =(r,d ,代表空间坐标和自旋;而 T 为 Wick 时序算符;y(xt) 、
y+(x't') 为海森伯绘景中场湮灭、产生算符; N 为 N 电子系统基态的正交归一波函数。
实际上,单粒子格林函数描述的就是 N 电子体系从N 1激发的动力学过程,对上述格林函数做傅里叶变换,可以得到另一个表达式:
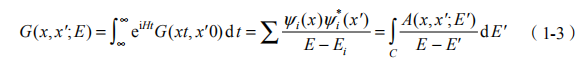
可见准粒子的本征值可由单粒子格林函数的极点确定。其中A(x,x',E)=
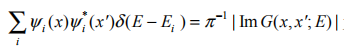
为谱函数,它对应着单粒子格林函数的虚部,反映准粒子的寿命。
求解准粒子的本征能量,我们可以构造如下运动方程:
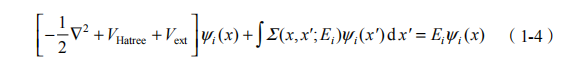
这一方程与 Kohn-Sham 单粒子运动方程非常类似,不同之处在于交换关联势Vxc 由自能算符 (x, x'; Ei ) 取代。求解这一方程需要做一系列的近似。准粒子本征值计算是在 Kohn-Sham 单粒子计算的基础上进行的,最关键的问题就是如何确定自能 。
原则上,人们可以通过 Hedin 方程组与 Dyson 方程联立求解自能 。考虑多体系统有一个外界的微扰d Vext ,采用简化组合坐标,我们可以定义不可约极化矢量
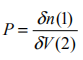 ,即势场总变化引起的粒子密度变化;顶角函数
,即势场总变化引起的粒子密度变化;顶角函数
,即势场总变化引起的粒子密度变化;顶角函数
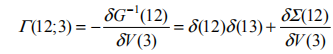
即势场总变化引起的倒易格林函数变化。Hedin 方程组可写为

逐次迭代求解屏蔽库仑势 W,最后即可求出自能 。注意G0 是无相互作用多体系统的单粒子格林函数,其形式与单粒子格林函数相同。
我们可以看到的是,从 Hedin 方程组出发的自洽求解方式过于复杂,人们常常采用一个简化的近似,取
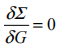 ,这样就可以丢掉顶角函数中的第二项,
,这样就可以丢掉顶角函数中的第二项,
,这样就可以丢掉顶角函数中的第二项,
即使 G (12;3)=d (13) 。这样一来,我们就可以得到自能的近似表达式:
(1,2)=iG(1,2)W (1+ ,2) ,简记为 =iGW ,此即所谓 GW 近似 [9](图 1-1)。实际上依据费曼图理论,GW 是自能 对屏蔽库仑势 W 的一阶展开,而高阶项对准粒子能量的影响很小,可以忽略不计,因此 GW 近似适用于绝大多数体系。
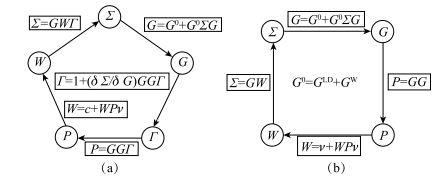
图 1-1 (a)Hedin 方程迭代法;(b)GW 近似迭代法。此图取自文献 [9]
计算自能 中极为重要的是计算屏蔽库仑势W ,而要计算屏蔽库仑势就必须准确计算介电常数 ,其满足的公式为
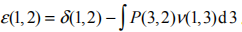 。介电常数 的计算是 MBGFT 及 GW 方法的核心任务,目前比较常用的方法
。介电常数 的计算是 MBGFT 及 GW 方法的核心任务,目前比较常用的方法
。介电常数 的计算是 MBGFT 及 GW 方法的核心任务,目前比较常用的方法
是无规相近似(RPA)[10]。在无规相近似下介电函数的倒空间矩阵元有如下形式:
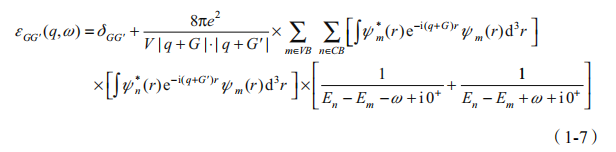
这部分的计算量十分惊人。在过去的 30 年里,人们提出了广义等离激元-极点模型 [11]、介电能带结构方法 [12] 以及静态介电函数模型 [13] 等试图简化计算, 但至今仍然很难在计算量与计算精度之间取得较好的平衡。以下是 GW 计算的基本步骤:
(1)从密度泛函理论计算所得基态波函数出发,构建单粒子格林函数 G;
(2)采取无规相近似,选取合适的介电常数模型近似,计算介电常数进而确定屏蔽库仑势 W;
(3)结合 G 与 W,利用 GW 近似计算自能 ;
(4)将以上所获得物理量代入 Dyson 方程,求解准粒子能量本征值;
(5)将能量本征值迭代回步骤(1),修正格林函数 G,如此往复,直至能量本征值收敛为止。
从上面的步骤我们可以看出,每一步更新格林函数和屏蔽库仑势都是一个非常麻烦的工作。但事实上,如此费劲的自洽计算并不一定能有利于提高计算精度。目前可靠的准粒子修正方法是所谓的 G W [14],即放弃上述步骤中的第五步,不进行自洽修正,并且不更新 G 与 W,是 GW 方法的零级近似。这种方法的有效性是有所保证的。密度泛函理论是一种平均场理论,而实际的有相互作用的多体系统会给这个平均场加上一个扰动。在GW 近似计算中, 我们可以把 -Vxc 视作对 Kohn-Sham 方程哈密顿量的一阶微扰。根据一阶微扰理论,准粒子的能量可以近似表示为
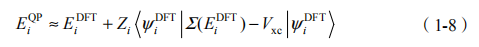
其中可定义重整化因子 Zi 为
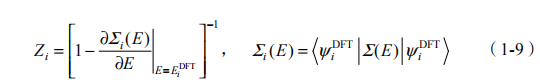
基于单粒子格林函数的 GW 近似方法可以有效地计算多体系统的基态和激发态,而多体系统中的激子行为则需要利用描述电子-空穴相互作用的Bethe-Salpeter 方程来处理。第一性原理计算中,我们用双粒子格林函数来描述激子的动力学过程,而它满足的运动方程就是 Bethe-Salpeter 方程(BSE)[15]。该方程的核心物理量是电子-空穴间的相互作用核 K,K 的吸引项耦合空穴与电子形成激子。通过求解BSE 方程,我们就能获得激子的能量、波函数、激发态寿命以及光学响应等性质。
首先定义双粒子的格林函数
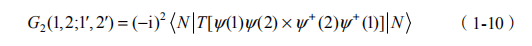
其中,我们仅考虑了电子- 空穴对的运动情况,即t1, t1'>t2 ,t2' 与t1, t1'<t2 ,t2' 两种情况。
其次,我们需要定义双粒子关联函数
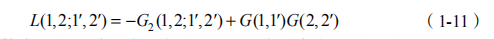
双粒子关联函数表示的是关联在一起的电子- 空穴对与相互独立的电子和空穴的差异。其运动满足 BSE 方程:
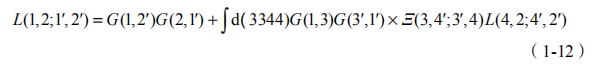
E是电子- 空穴相互作用核,也被称为 Bethe-Salpeter 核。我们也可以相应地用双粒子关联函数来定义系列物理量:极化率 P(1, 2)=iL(1, 2;1+, 2+) ;介电函数
(w)=1-vcp(w)以及相互作用
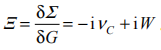
程求解手续如图 1-2 所示。
值得注意的是,通常在求解BSE 方程中有两个重要近似,一是获得倒易介电常数时采用的无规相近似;二是把 BSE 哈密顿量中共振跃迁与反共振跃迁的耦合项做微扰处理的 Tamm-Dancoff 近似 [16]。此外,屏蔽库仑势 W 也常常被静态处理 [17]。
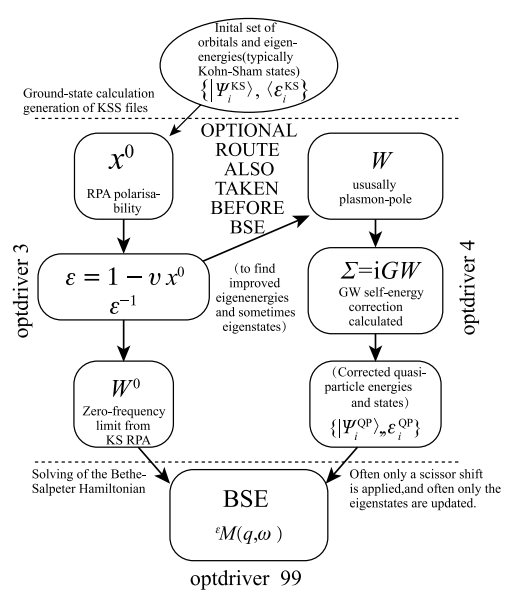
图 1-2 典型的 BSE 方程求解手续,本图取自 Abinit 网站
如图 1-3 所示,以格林函数为基础的 GW 方法与 Bethe-Salpeter 方法原则上精确描述了有相互作用多体体系的基态与激发态,修正了密度泛函理论带来的带隙低估等尴尬,在准粒子能量、寿命、光学响应上获得了与实验相当符合的理论预言,其研究对象也被扩展到半金属 [18]、点缺陷 [19] 以及关联电子材料等半导体体系 [20]。尽管其计算量巨大,但仍展现出了广阔的前景和强大的生命力。
近年来人们采用基于含时密度泛函(time-dependent density function theory,TDDFT)[21] 和 Ehrenfest 定理来描述电子波函数实时演化及和原子振动相耦合的计算方法 [22]。该方法可用来研究较大尺度上半导体和纳米材料电子激发态的光吸收和电子动力学过程。
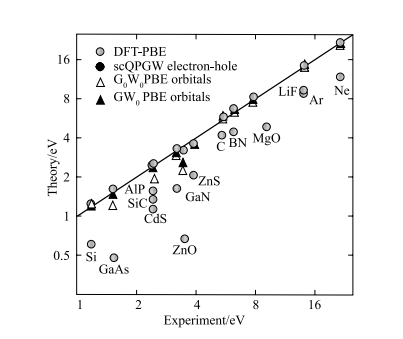
图 1-3 常见半导体材料中各 GW 算法与实验、DFT-PBE 之比较。取自文献 [19]
根据量子力学原理,原子核-电子体系的演化满足含时薛定谔方程:

其中,是普朗克常量; 是体系的波函数;rj 是第 j 个电子的坐标;RJ 是第J 个原子核的坐标。整个体系的哈密顿量包含电子动能、原子核的动能、电子间的库仑斥力、原子核间的库仑斥力、电子-原子核间的库仑吸引和外势场 Uext 的贡献,写为
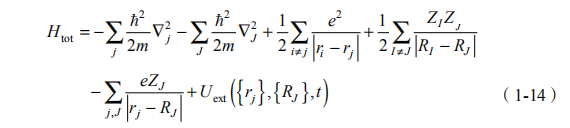
在实际计算中,电子的运动可以用含时密度泛函理论描述,而原子核则采用经典的描述,并取原子核密度分布为 rJ (R, t) = d (R - RJ (t)) , ,得到电子和原子核的运动方程分别是
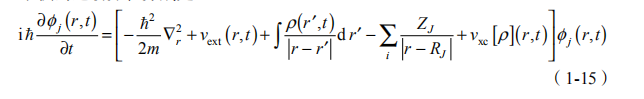
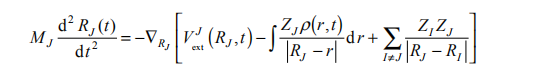
其中,j是单粒子近似下的电子 Kohn-Sham 轨道; 是电子的总密度;vxc 是电子的交换关联泛函;MJ 和 ZJ 分别是第 J 个原子核的质量和电荷。
在密度泛函理论优化得到的原子结构基础之上,先进行基于玻恩-奥本海默近似的分子动力学模拟;在某一时刻,改变电子占据态使一部分电子占据在空态并在原能级产生相应的空穴来模拟激发态电子的占据情况,然后使用含时密度泛函理论演化整个体系所有能级上的电子波函数,同时计算新的波函数所对应的原子受力并获得原子下一时刻的位置和速度。重复这一流程,就得到半导体和纳米体系激发态上的电子-离子实时演化的动力学过程。该方法的优点在于使用:①有限空间大小的局域原子轨道做波函数基失;②实时进行波函数演化。这些可导致运算时间随着体系中原子数目呈线性增长,而不是现在的成指数或者三次方增长,因而适合于研究纳米材料和有机体系光吸收、能量转化等激发态现象 [23]。这种方法已被应用于染料太阳能电池中电子注入动力学、电子复合动态、表面光催化、二硫化钼等二维体系的界面电子动力学模拟等方面。
在能带计算中,最基本的近似是玻恩-奥本海默近似 [24]。它将固体中电子和晶格的运动退耦,从而分别求解这两个子系统的能谱。目前存在两种计算方案,即同时处理电子和离子的动力学(如 Car-Parrinello 方法)或分别处理电子和离子动力学(如玻恩-奥本海默近似)。在这两种方法中,电子的动力学部分均在量子力学框架内处理。对于玻恩-奥本海默近似,在大多数固态材料中(如离子键和共价键体系)是非常好的近似。在氢键系统中,由于原子核质量较小,电子和原子核之间的耦合变得十分重要。在这种情况下, 玻恩-奥本海默近似的结果值得检讨。
有趣的是,玻恩-奥本海默近似也可以用来分析单粒子的动力学行为。人们发现快运动可以对慢运动诱导出规范场,这点最早由 Wilczek 发现。通常这类规范场并没有导致特别的效应,但对于某些特别的体系,如存在自旋轨道耦合的体系,则可以诱导出自旋依赖的、洛伦兹力型的规范场,导致自旋和量子自旋霍尔效应。
依据 Hellmann-Feynman 原理,电子系统给每个原子核一个特定的力。这个力加上原子核之间经典的库仑作用,给出的是每个原子核在这样一个特定构型下受到的总体的力。连续地变换原子核的构型,进而连续地变换这一系列分立的静态总能,以及原子核本身的受力情况。当原子核的构型走遍整个原子核系统的高维构型空间的时候,这些静态能就会给出一系列分立的高维曲面,即玻恩-奥本海默势能面。通常人们认为在这些势能面上,电子的行为是量子的,而原子核本身的行为是静态的、经典的。
在此基础上,为了能够进一步引入原子核行为的量子描述,最直接的方式就是从这些玻恩-奥本海默面出发,建立原子核运动的薛定谔方程并求解, 进而得到相关的统计(甚至动力学)性质。但对于大多实际系统,由于计算量的原因,以上方法失效。为解决困难,人们发展了路径积分分子动力学方法。路径积分分子动力学方法的出发点是量子力学的路径积分表述 [25-27]。在这个表述中,描述量子世界中两个事件之间关联的最基本的物理量就是传播子,它的形式可以写为
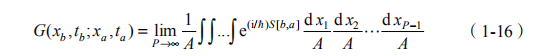
也就是说,传播子等于它所描述的两个时间 a 与 b 之间的所有路径的贡献的和。每个路径的贡献有一个权重,是由它所对应的 Action,也就是上式中的S[b,a] 决定的,S 等于拉格朗日量在这两个事件间的积分。A 是一个归一因子,为
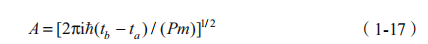
在以上基础上,可以构造一个量子系统的密度矩阵,它决定这个量子系统的所有统计性质:
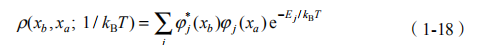
然后依照传播子在波函数表述与路径积分表述中的等价性,将密度矩阵在路径积分下的表述形式写成 [28]
自 20 世纪 80 年代开始,这种路径积分数值方法与分子动力学/蒙特卡罗的结合使得它在分子模拟领域就原子核量子效应的描述上,从方法论的发展以及应用的角度都得到了巨大的发展。其中,Chandler 和 Wolynes 于1981 年的工作 [29] 为虚时间路径积分和分子动力学相结合开辟了道路。此后,Berne 等先后对该方法进行了一定程度的扩展 [30]。路径积分分子动力学的研究应该说是路径积分数值方法与分子模拟手段较早的一个结合。与这些路径积分分子动力学方法的发展几乎同步,Ceperley 和 Pollock 也将路径积分数值方法与蒙特卡罗方法进行了有效的结合,并对液体 He4 在低温下的一些统计行为(包括超流性质)进行了较为严格的数值模拟 [31,32]。
对温度的虚时处理,使得人们可以在分子模拟中就统计性质而言将原子核的核量子效应与热效应同时进行处理。然而,路径积分分子动力学/蒙特卡罗模拟依赖于原子核之间相互作用的描述,即力场模型。虽然力场模型是基于精确的第一性原理电子结构计算或实验结果的,但是在一些特别的系统中,如可能发生化学键断裂现象的系统中,力场方法往往会失效。为了解决这个困难,自 20 世纪 90 年代,Tuckerman、Marx、Parrinello、Klein 等将基于第一性原理电子结构计算的 Car-Parrinello 分子动力学方法与路径积分分子动力学方法进行了有效的结合 [33,34],并利用该方法对一些氢键系统中的质子传输进行了系统的研究 [35]。2000 年以来,基于玻恩-奥本海默分子动力学方法的路径积分分子动力学研究也逐步展开,比如李新征和 Michaelides 等在这个方向上做的一些工作 [36,37]。为减少数值上很大的计算量,最近,由 Ceriotti 等提出的量子热浴方法在数值上提供了一个有效的方法 [38]。总之,基于路径积分方法的分子模拟手段已经成为目前人们在真实体系中针对核量子效应的统计性质展开研究的一个成熟的、重要的工具,使得人们可以在原子尺度对与核量子效应相关的诸多物理、化学性质进行深入的分析。
但是,紧束缚方法在许多情况下,比如计算体系的尺度比较大、模拟长时的物理问题、综合的结构搜索等,依然具有不可替代的优势。这主要是因为紧束缚方法具有更好的计算效率,速度上比 DFT 快几个数量级。密度泛函紧束缚(DFTB)方法[39] 是在Slater-Koster 两中心积分近似[40] 的基础上, 通过拟合 DFT 的计算结果得到参数化跃迁函数,从而产生类似 DFT 计算的结果。因此,与其他紧束缚模型相比,DFTB 方法具有较好的精度和可移植性,不仅适用于共价体系,而且适用于金属体系,如金团簇 [41],甚至还可以用来处理具有强关联作用的体系,如铁团簇等 [42],在材料计算领域具有比较广泛的应用。目前,已有的跃迁参数表不仅包括单元素体系(如碳、硅等 [39,43]),也包括多元素体系(如硼氮、过渡金属硫化合物等 [44]),具体可见 dftb.org。
DFTB 方法的核心是通过 DFT 计算获得跃迁参数表。
(1)定义类似原子轨道的基函数(由 Slater 轨道和球谐函数组成),
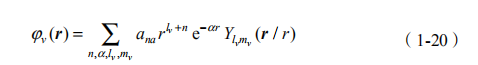
然后,求解 Kohn-Sham 方程:
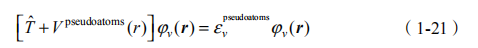
(2)Kohn-Sham 方程中势能项定义为
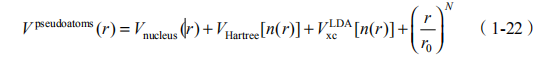
(3)包括:原子核势能 Vnucleus、电子排斥作用形成的有效势 VHartree 和局域密度近似下的交换关联作用V LDA 。额外项(r/r )N 用来加速波函数的收敛,起到压缩电子密度的作用。N 一般取到 2。rcov 定义为原子的共价半径,
r0=2rcov。
通过自洽求解 Kohn-Sham 方程,得到基函数jv (r,我们就可以通过它来获得紧束缚模型的参数。重叠积分矩阵元是基函数之间的积分,是原子间距的函数。为了得到哈密顿矩阵元,首先需要构造一个有效势函数:
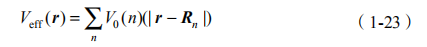
其中, Rn 是原子的位置矢量;V0 是这个原子的 Kohn-Sham 势,也就是公式(1-22),但是去掉最后一项(r/r )N。有了有效势函数,哈密顿矩阵元就得到了:
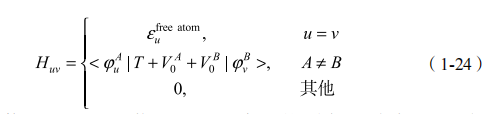
原子波函数和 Kohn-Sham 势以 n 和 n′ 所标示的不同原子为中心。不难看出,哈密顿矩阵元仅仅包括两中心积分。哈密顿矩阵的对角项取自由原子的本征值free atom ,保证了孤立原子能量极限的合理性。
DFTB 的能带计算就是以得到的重叠积分和哈密顿矩阵元(跃迁积分) 为基础进行的。与其他紧束缚模型一样,DFTB 的总能包括电子能带能量和一个短程的二体排斥势能 Erep:
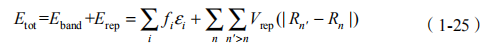
其中,Vrep 描述原子核之间的排斥作用。但是,实际中 Vrep 描述的是 DFT 电子能带与紧束缚能带 i 的差,在一定程度上包含更为复杂的相互作用。fi 为第 i 轨道的电子占据数。
以上讲述了 DFTB 方法的基本思路。在这个模型的基础上,可以继续发展电荷的自洽场计算 [45](scc-DFTB)以及对于电子自旋极化效应的计算 [46]。电荷的自洽场计算指为了提高原子之间电荷转移量定量描述而进行的自洽计算,从而提高 DFTB 的计算精度。实际上,已经有实例证明 scc-DFTB 在精度上与 DFT 相当 [47]。scc-DFTB 提供共线和非共线两种处理电子自旋极化的方法。图 1-4 的结果是在计算由碳、氧、氮、硅、硫等元素组成的分子的超精细耦合常数时,DFT 计算结果和 scc-DFTB 计算结果与实验的比较 [48]。可以看出,绝大多数 scc-DFTB 数据与实验一致。同时,scc-DFTB 结果与 DFT 结果也基本一致。
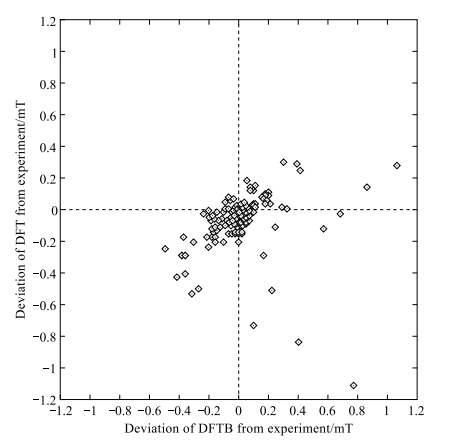
图 1-4 以实验为参照,DFT 计算误差分布和 scc-DFTB 计算误差分布。0 mT 标示计算与实验完全一致。摘自文献 [47]
同时,含时 DFTB 方法 [49]、基于 DFTB 的格林函数方法 [50]、输运性质的计算 [51] 以及对范德瓦耳斯相互作用的处理 [52],丰富了 DFTB 方法的用途。
第三节 半导体低维体系中的拓扑量子态
根据拓扑不变量对电子态划分是一个强有力的工具,最先由 Thouless、Kohmoto、Nightingale 和 den Nijs (TKNN)[53] 在整数量子霍尔效应中尝试。他们确立了非相互作用的整数量子霍尔效应相应的拓扑不变量。TKNN 整数n 给出了每个带的量子霍尔电导率 =ne2/h,n 由布洛赫波函数对磁布里渊区的积分给出,对应于环上 U(1) 主纤维丛的第一陈类。这种拓扑分类区分了普通绝缘体和量子霍尔态,并且解释了霍尔电导率对弱无序和无作用的不敏感性。非零 TKNN 整数同样与样品边界出现的无能隙边缘态密切相关。
自从量子霍尔效应发现以来,人们一直在时间反演对称的系统中寻找拓扑量子态。Haldane 最早在这方面做出开创性的工作,即无朗道能级的量子霍尔效应模型 [54]。他采用二维的蜂巢晶格模型(即类石墨烯的晶格),考虑相邻晶胞通过大小相等、方向相反的磁通,显示出时间反演对称的系统在没有外磁场的情况下霍尔电导率 xy 出现非零的量子化;在临界参数下有无质量费米子产生,并且展示出 2+1 维所谓的“宇称反常”;采用模型的低能量态模拟了“2+1 维”相对论性量子场论,显示出所谓的“宇称反常”和 2+1 维手性费米子的特性,如图 1-5 所示。
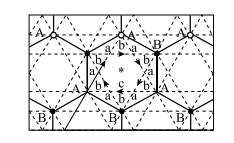
图 1-5 Haldane 模型
Haldane 采用的是“二维石墨”(即石墨烯)模型。二维石墨有蜂窝状结构,由两个互相穿透的三角晶格(A 子格和 B 子格)构成,在元胞中各有一个格点。他采用每点含一个轨道和一个相邻不同子格格点间跃迁的实矩阵元 t1 的紧束缚模型,并考虑破坏空间反演性的位能(A 格点为 +M;B 格点为-M)。这种模型有 C6v (M=0) 和 C3(M ≠ 0) 的点群对称性。该模型中时间反演不变。Haldane 在次近邻点中引入第二项实跃迁项(即相同子格的最近邻点)。尽管它破坏了原来模型中的粒子空穴对称性,但这并不改变空间群对称性。为了保证时间反演不变,Haldane 在垂直于二维平面的 z 方向加入局域周期性磁通密度 B(x),同时保持晶体的完全对称性和穿过元胞总磁通量为 0。因为元胞的净磁通消失了,矢势 A(r) 可以选为周期性的。这种局域场的效应是将不同点间的跃迁矩阵元乘以幺模的相因子
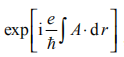 ,其中积分沿跃迁路径,取作直线。总相位沿闭合路径累加即为以单位量子磁通
,其中积分沿跃迁路径,取作直线。总相位沿闭合路径累加即为以单位量子磁通
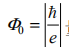 量度的(量子化的)磁通。因为第一近邻跃迁的闭合路径包含了整个单位元胞(因此没有净通量),t1 矩阵元不受影响。t2 矩阵元获得一个相位f = 2p(2Fa + Fb ) / F0< 其中Fa 、Fb 为通过元胞中 a 和 b 区域的磁通。次近邻跃迁方向如图 1-5 中箭头所示,其强度为t2 exp(+ i f) ,,可以看到在局域磁场存在下哈密顿量获得了手性。这样一个内建磁场的来源原则上可以用被放置于六边形原胞(Wigner-Seitz)中间的磁偶极矩 来实现,磁场 B(r) 是偶极场的总和。
量度的(量子化的)磁通。因为第一近邻跃迁的闭合路径包含了整个单位元胞(因此没有净通量),t1 矩阵元不受影响。t2 矩阵元获得一个相位f = 2p(2Fa + Fb ) / F0< 其中Fa 、Fb 为通过元胞中 a 和 b 区域的磁通。次近邻跃迁方向如图 1-5 中箭头所示,其强度为t2 exp(+ i f) ,,可以看到在局域磁场存在下哈密顿量获得了手性。这样一个内建磁场的来源原则上可以用被放置于六边形原胞(Wigner-Seitz)中间的磁偶极矩 来实现,磁场 B(r) 是偶极场的总和。
,其中积分沿跃迁路径,取作直线。总相位沿闭合路径累加即为以单位量子磁通
量度的(量子化的)磁通。因为第一近邻跃迁的闭合路径包含了整个单位元胞(因此没有净通量),t1 矩阵元不受影响。t2 矩阵元获得一个相位f = 2p(2Fa + Fb ) / F0< 其中Fa 、Fb 为通过元胞中 a 和 b 区域的磁通。次近邻跃迁方向如图 1-5 中箭头所示,其强度为t2 exp(+ i f) ,,可以看到在局域磁场存在下哈密顿量获得了手性。这样一个内建磁场的来源原则上可以用被放置于六边形原胞(Wigner-Seitz)中间的磁偶极矩 来实现,磁场 B(r) 是偶极场的总和。
采用两个子格布洛赫波函数构造的二分量旋量为基,可以使哈密度量对角化。哈密顿量对角化后可以写为
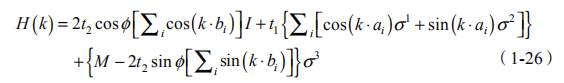
其中,a1,a2,a3 为 B 格点到最近邻 3 个 A 格点的格矢。布里渊区为相对Wigner-Seitz 原胞转动 90 后的正六边形晶胞。从上式可以得出,仅当 3 个 Pauli 矩阵的系数均为衰减时带隙才会闭合。
当费米能级处于带隙中时, xy 在零温时是量子化的,它的值可通过
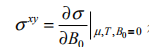 得到, 是二维电荷密度,B 表示 z 方向均匀外磁场的磁通密度。将哈密顿量在 k 0 附近展开d k = k - k 0 , hd k P x , y) ,可以得到两个顶点附近两个独立的哈密顿量:
得到, 是二维电荷密度,B 表示 z 方向均匀外磁场的磁通密度。将哈密顿量在 k 0 附近展开d k = k - k 0 , hd k P x , y) ,可以得到两个顶点附近两个独立的哈密顿量:
得到, 是二维电荷密度,B 表示 z 方向均匀外磁场的磁通密度。将哈密顿量在 k 0 附近展开d k = k - k 0 , hd k P x , y) ,可以得到两个顶点附近两个独立的哈密顿量:
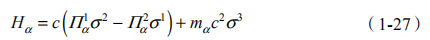
进而得到能谱。从上式可以看出,第一项非常类似于 Rashba 自旋轨道耦合项,第二项代表质量项,它打开能隙。
(1)对于 B0=0,相对论性朗道能级为
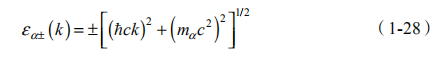
其中,
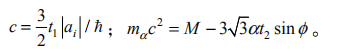
(2)对于 B0 ≠ 0,有
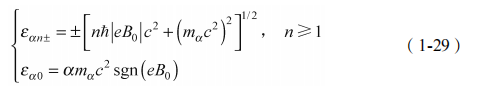
在外净磁场非零情况下,每个 n ≥ 1 由上面的带产生的朗道能级都被下面的带产生的朗道能级抵消(B0 -B0 变换下是对称的)。然而,n=0 的零模式能量在变换下不是对称的:如果从上面能带产生取正号,从下面能带产生取负号,则时间反演不变导致 xy=0。
为了计算模型中 m 和 m 反号时的 xy,连续地在外磁场存在的情形下改变 m+ 和 m-直到它们变得相等,与此同时变化费米能级使得它始终处在带隙中。通过这种方式得到的朗道能级占据数和通过连续改变时间反演不变系统的外场获得的占据数相比较,会显示出它们在完全占据一个朗道能级上的差别。因此,在零温下,同时有一个固定化学势时,向 m+ 和 m-反号的系统施加外加弱磁场会有一个额外的场依赖的基态电荷密度 = e2B /h(当 m 和m-同号时,其和场独立的电荷密度相关)。
这允许 xy在 B =0 时用 ve2h 衡量, 其中
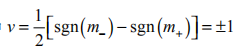 或 0。不考虑自旋的电子 v 为 M / t2 和 的函数的模型的相图如图 1-6 所示。
或 0。不考虑自旋的电子 v 为 M / t2 和 的函数的模型的相图如图 1-6 所示。
或 0。不考虑自旋的电子 v 为 M / t2 和 的函数的模型的相图如图 1-6 所示。
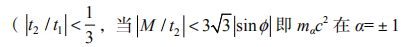 反号时,零场量子霍尔效应的相会出现)。
反号时,零场量子霍尔效应的相会出现)。
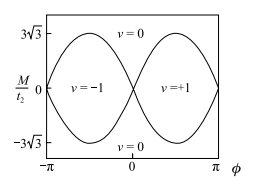
图 1-6 无自旋电子 |t2/t1|<1/3 时的相图
当 m =0 时,由两个无能隙顶点哈密顿量导出的费米场理论有电荷共轭对称性(粒子空穴对称性),这在推导出该哈密顿取t2 ≠ 0时的晶格模型中并不存在。在连续场理论中充满电子态的费米海没有下界, xy 的绝对值而非相对值是不确定的。Jackiw[55] 主张顶点哈密顿量的电荷共轭对称性和 m =0赋予 xy=0 是在电荷空穴对称朗道能级的情况下, B0 1 0 的零模朗道能级是 半填充时。这暗示了当零模式是填充时,量子霍尔系数 v = (1/2)a ,而在空态v = (1/2)a 这就是提议了“电荷分数化”,而违背了一个非相互作用电子系统只允许有整数量子霍尔效应的事实。这里研究的模型显示高能量怎样肢解了由顶点相对论性哈密顿描述非倍增费米子的模型结构,该哈密顿在低能下必须破坏电荷共轭对称性,从而将额外的 1/2 给 v,产生整数的量子霍尔效应。因此,尽管低能能谱是由非倍增手性费米子构成的,但它们的共轭子必须在高能态产生合理的整数量子霍尔效应。
当电子自旋包含在内而没有其他改变时,两个自旋取向有相同的贡献, 从而 xy 变为了 2 倍。然而,一个有完全晶格对称性的周期性局域磁场也会以一个塞曼项耦合到电子上H′= S z,其中 S z 是方位角的电子自旋角动量。这一项相对以gf S z 能量间隔错置了自旋向上和自旋向下的带,而且如果间隔超过了费米能级的能隙,系统会变成部分自旋极化的金属。如果
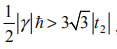
量子霍尔相会被完全消除,但如果该项小一些,相会在足够小的 M 和 t2sinf 下保持(随 M 改变,从常规到反常半导体相的直接相变会被插入其中的自旋极化金属相替代)。对于之前提到内建磁场的实现,g ?(在里德伯单位下)由C ′g/a2 给出,C ′ 是序单位的另一个几何参数,g 是朗德因子。
尽管这里展示的特殊模型在物理上很难直接实现,但它暗示,原则上量子霍尔效应可以在更广泛的时间反演不变性被破坏的相关现象中出现,并且不必要求外磁场的存在,而可以在准二维系统中由磁序产生。
第四节 Z2 拓扑序和量子自旋霍尔效应
在 Haldane 的工作基础上,2005 年 Kane 和 Mele[56] 提出了在石墨烯中实现量子自旋霍尔相。他们发现在时间反演不变的有体态能隙的电子体系中存在无能隙的金属边缘态。他们发现这个相与 Z2 拓扑不变量 [57] 有关,使其与普通绝缘体相区别。这种在时间反演不变的哈密顿量中定义的 Z2 分类和量子霍尔效应定义的陈数分类相似。对于这些物质的态,可以根据它们的拓扑性质进行区分。如同整数量子霍尔效应由拓扑整数 n 来表征,这确定了霍尔
第一章 半导体能带理论
电导率量子化取值和手性边缘态的数目。量子自旋霍尔相和分数量子霍尔相一样,是一个新的拓扑可分辨的物质态。
他们计算了石墨烯两带模型的量子自旋霍尔效应,并提出一个适用于多带和相互作用系统的一般性的讨论。由于石墨烯由碳原子构成,并且碳原子的原子序数较小(Z=4),因此自旋轨道耦合很弱(自旋轨道耦合强度正比于 Z 4)。作为一个模型,石墨烯定义了新的一类自旋霍尔绝缘体,提供了寻找其他强自旋轨道耦合的拓扑绝缘体的全部物理基础。
考虑最简单的石墨烯紧束缚哈密顿量(一个 电子含有平面镜像对称的紧束缚模型),其中计入时间反演不变的自旋轨道相互作用:

其中,第一项是晶格中的最近邻跃迁;第二项是镜像对称的自旋轨道相互作用;第三项是来自垂直电场的 Rashba 自旋轨道相互作用;第四项是交错子格势。自旋轨道耦合 z zsz 产生的能隙与交错子格势 z 或者 z z 形成的带隙是不同的。后者的基态与强耦合下的普通绝缘相(其中两个子格退耦合)绝热联系。自旋轨道耦合对应的哈密顿与 Haldane 模型有联系,sz= 1 各自单独考虑时破坏了时间反演对称性,与 Haldane 模型中的无自旋电子相等价,可以通过引入没有净磁通的周期性磁场来实现。
他们发现,自旋轨道相互作用在有体态能隙的单一石墨烯平面会导致时间反演不变的量子自旋霍尔(QSH)态,表现为在样品边界处出现一对无能隙自旋分离的边缘态。当 Sz 不变时,石墨烯和一个简单绝缘体的区分容易理
解:每种自旋有独立的 TKNN 整数 n- , n 。时间反演对称性要求 n- + n = 0 , 但是二者差值n- - n 非零并定义了一个量子自旋霍尔电导率。在能隙中会导
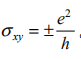 致量子化的霍尔电导 xy空间的拓扑陈数。
致量子化的霍尔电导 xy空间的拓扑陈数。
致量子化的霍尔电导 xy空间的拓扑陈数。
尽管由于带间耦合(如 Rashba 自旋轨道耦合),镜像对称破坏,或是无序导致 Sz 不再守恒,这些扰动会破坏自旋霍尔的电导率量子化,但不会破坏QSH 的拓扑序,因为Kramers 定理阻止时间反演不变的扰动在边缘打开能隙。因此,QSH 基态和简单绝缘体是可区分的。从石墨烯模型出发显示,尽管 Sz 不恒定,QSH 相却是鲁棒的。
根据布洛赫定理,H 可以对角化,以布洛赫波函数 |u (k)>洛赫哈密顿量为
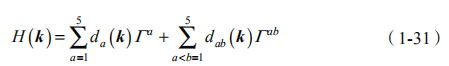
其中,d 矢量和 d 张量由上面 4 项系数表示。由于 H(k+G)=H(k),因此 H(k)是定义在环上的。上述哈密顿量给出四条能带,其中两条被占据。
对于lR =< 0 ,存在大小为6 3lSO - 2lv 的的能隙。 lv > 3 3lSO 能隙取决于 lv ,且该系统为绝缘体。3 3lSO > lv 描述了 QSH 相。尽管Rashba 项(第四项)破坏了 Sz 守恒,对于lR < 2 3lSO ,相图中有限区域与lR = 0 的 QSH 相之间存在绝热联系。图 1-7 显示了通过解锯齿形(zigzag)条带的几何对应的晶格模型中的属于绝缘和 QSH 相中的代表点得到的能带图像。两种相都有体态能隙和边缘态,QSH 相中的边缘态成对横穿能隙。
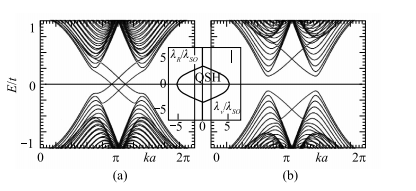
图 1-7 一维锯齿形条带的能带:(a)QSH 相 v=0.1t;(b)绝缘体相 v=0.4t。二者均取 SO=0.06t 和 R=0.05t。边缘态交叉于 ka=
这些边缘态的行为显示了两种相之间明显的区别。在 QSH 相中每个边缘,体态能隙中的每个能量取值都有一个时间反演的相伴的本征态。因为时间反演对称性阻止了 Kramer 简并态的混合,所以这些边缘态对于微扰是鲁棒的。这些无能隙态甚至可以存在于空间反演进一步破坏的情况下(如在哈密顿量中去掉 C3 转动对称性)。由于单粒子弹性背散射是被禁戒的,因此弱无序不会导致这些边缘态的局域化。
时间反演对称性确定了布洛赫哈密顿量 H(k) 张成空间两个重要的子空间和其对应的占据态波函数 |ui (k )> 。“偶”子空间,满足Q H k )Q -1 = H k ) ,具有|Q ui (k )>在 U(2) 转动下等价于|Q ui (k )>的性质。根据对角化后的哈密顿量形式,这个子空间 dab(k)=0。时间反演对称性要求 H(k) 在G 点和 M 点上属于偶子空间,如图 1-8 所示。奇子空间有基|ui (k )> 张开的空间与基|ui (k )> 张开的空间相正交的性质。我们通过奇子空间的一系列 k 来研究 Z2 分类。

图 1-8 QSH 相 k* 点上的 P(k) 零点:(a) ≠ 0;(b) =0;(c) 实线表示 QSH 相,虚线表示绝缘相;(d)实心表示对称点不含零点,C 为 Z2 指标定义中的积分路径
特殊的子空间可以通过考虑交叠矩阵 |ui (k ) Q u j (k ) >确定。由Q 性质,可以清楚地看到矩阵是反对称的,并且可以用单复数 eij P (k ) 表达,实际上等于 Pfafian:
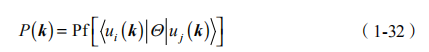
对于 2*2 反对称阵 Aij,简单地会对 A12 选择。我们在下面看到 Pfafian
是多于两个占据带时自然的推广。P(k) 不是规范不变量。在 U(2) 变换下= Uij ui, P' = P e2iq 。在偶子空间中, ui在 U(2) 转动下与 ui等价,
我们有 |P (k )| = 1 。在奇子空间中 P(k)=0。
如果没有空间转动限制它的形式,P(k) 零点通过调制两个参数可以出现,并通常出现在布里渊区的点上。一阶零点产生在由相反“涡量度”的时间反演的点 k* 构成的对上,P(k) 的相位在点附近沿相反方向增加。对于 v ≠ 0,QSH 相可以通过 P(k) 的单对一阶零点与普通绝缘体相区别。我们模型的 C3 转动对称性限制了 k* 在布里渊区的顶角上。如果不存在 C3 转动对称性,k* 可以产生于除了四个对称点 |P(k)|=1 之外的任何地方。零点对数是 Z 拓扑不变量。这可以通过两个对 k* 放到一起后当k* =- k* 时相消看出。但是单一的 k* 零点对在 点或 M 点遇上时不会相消,因为 |P(k)|=1。
如果时间反演被破坏,那么零点将不再被阻止相消,QSH 相的拓扑分辨将会丢失。
因此,Z2 指标可以由 P 的复零点对数决定。这可以通过计算 P(k) 的相位沿围绕半个布里渊区闭合路径的卷绕(winding)得到(定义使 k 和-k 不同时包含)。

其中,C 是图 1-8 中所示的路径。
当 v=0(如在石墨烯中)时,H 有 C2 转动对称性,当其与时间反演结合时限制了 H(k) 的形式,并允许 P(k) 取实。P(k) 的零点之后会出现在线上, 而不是在点上。我们发现在绝缘相中零点并不存在,但是在 QSH 相中会被封入 M 点。这种情形下我们发现 Pfaffian 定义的 P(k) 也决定了 Z2 指标(由C 路径上符号变化次数的一半给出),只要我们把收敛因子 也包括在内。需要指出的是,I 的符号取决于 的符号,而 I 对 2 取余则不是。我们可以总结QSH 相和绝缘体用 Z2 指标 I 区分。
考虑到重原子的自旋轨道耦合较强,张首晟等 [58] 独立地提出在 HgTe- CdTe 半导体量子阱中实现量子自旋霍尔效应。当量子阱的宽度增加时,电子态在临界宽度 dc 出现一个从正常能带序向能带反转体系的转变。他们发现, 这种转变对应一个从普通绝缘体相到有一对手性(螺旋性)边缘态的 QSH 相的拓扑量子相变。
他们认为,时间反演对称性在手性边缘态的动力学中扮演了重要角色。当偶数对边缘态在边缘时,杂质散射和多体相互作用会在边缘打开一个能隙,并使系统变为拓扑平庸。然而,当有奇数对手性边缘态在边缘时,这些效应不会打开能隙,除非时间反演对称性在边缘被自发破坏。
他们研究了 型 HgTe/CdTe 半导体量子阱,发现 QSH 态可以在能带反转的区域实现。他们在一般的对称性考虑基础上,利用标准的半导体能带扰动模型(即 k p 理论),发现 点附近的电子态可以用 2+1 维狄拉克方程描述。垒区材料 CdTe 有正常的能带序列,s 型 6 带位于 p 型 8 带的上方,阱区材料 HgTe 有反转的能带序列, 6 带位于 p 型 8 带的下方。两种材料都是直接能隙,位于布里渊区 点。他们忽略了体态分裂的 7 带,采用了 6 带Kane 模型:
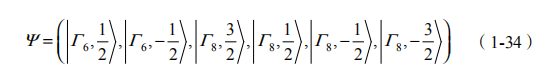
在 [001] 方向生长的量子阱,立方和球对称性退化为平面内绕轴(z)转动对称性。六带结合形成 3 个量子阱子带,即 E1、H1 和 L1 自旋向上和自旋向下的态 。L1 子带与其他两个带分离,我们忽略它,从而得到有效 4 带模型。用基
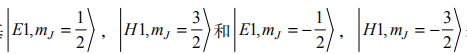 表示。
表示。
表示。
上式中 d3(k) 中包含了 E1 和 H1 的能量差,是一个重要的参数,即质量或者能隙常数 M。在量子阱几何中,HgTe 能带反转会导致 HgTe 层取特定厚度 dc 时发生能带交错。d< dc 是通常情况,对于 d >dc,E1 和 H1 能带必须在某个dc 处交叉,M 在转变点 d=dc 的两侧取相反符号。采用 点附近紧束缚模型可以使有效哈密顿进一步简化。霍尔电导率由
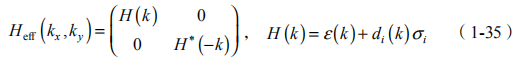
上式中 d3(k) 中包含了 E1 和 H1 的能量差,是一个重要的参数,即质量或者能隙常数 M。在量子阱几何中,HgTe 能带反转会导致 HgTe 层取特定厚度 dc 时发生能带交错。d< dc 是通常情况,对于 d >dc,E1 和 H1 能带必须在某个dc 处交叉,M 在转变点 d=dc 的两侧取相反符号。采用 点附近紧束缚模型可以使有效哈密顿进一步简化。霍尔电导率由
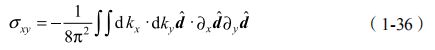
他们讨论 QSH 态的实验探测。他们提出采用纯电学测量可以探测 QSH 态的基本特性。通过扫描栅极电压,我们可以测量两末端电导率 GL,R,从 p掺杂到体态绝缘再到 n 掺杂区域。在体态绝缘区域,对于 d<dc,普通绝缘体低温下GL,R 消失,然而对于 d >dc,GL,R 会非常接近 h 。
张首晟等还提出利用 InAs/GaSb 量子阱实现量子自旋霍尔效应 [59],目前已被美国莱斯大学的杜瑞瑞小组 [60] 实验证实。该体系是由两种正常能带结构的半导体 InAs 和 GaSb 构成的,由于两者之间特殊的能带相对位置,可以形成两者之间的能带翻转,从而实现拓扑转变。
常凯等 [61] 提出,利用极性界面形成的强极化电场可以在主流半导体材料(如 InN/GaN,Ge/GaAs)极性界面异质结中实现拓扑相。他们通过计算表明,在极性界面处存在高达 10 MV/cm 的局域电场,该电场可以显著地改变量子阱区材料的能隙,改变的范围可达 1 eV 量级。这主要是源自量子限制效应和局域强极化电场的竞争。随着层厚的增加,电场效应开始显现,导致能隙急剧下降,并形成翻转的能带序。目前来自美国 AMES 国家实验室和Sadia 及普林斯顿大学的实验组 [62] 证实了局域电场的存在,理论计算结果和实验吻合。
三元Heusler 化合物化学式为X2YZ 或XYZ(X、Y 为过渡或者稀土金属, Z 为主族元素),它们形成了一类非常适合于自旋电子应用的材料。这些化合物的半导体属性来自其强烈的共价成键倾向。从基本结构和成键考虑,有 18 或 24 个价带电子的 X2YZ Heusler 化合物(L21 结构)和有 18 个价带电子的XYZ(C1b)预期在费米能上有带隙。很多 C1b 结构的 Heusler 化合物是三元半导体,在结构上和电子学上与二元半导体有关,并且可以调制带隙的大小。通过选择有合适组分的化合物实现所需要的能带反转以及自旋轨道耦合的强度。基于第一性原理计算,Chadov 等证明了大概有 50 种 Heusler 化合物会表现出和 HgTe 相似的能带行为 [63]。这些零带隙半导体拓扑绝缘态可以通过施加应力或者设计量子阱结构来实现。很多三元零带隙半导体(LnAuPb、LnPdBi、LnPtSb 和 LnPtBi)包含稀土元素 Ln,可用于实现超导电性(如LaPtBi)到磁性(如 GdPtBi)和重费米子行为(如 YbPtBi)。
特别是化合物,如 YPtSb、YPdBi 和 ScAuPb,它们晶格常数的实验值和普通绝缘体与拓扑绝缘体临界值接近,且所有相关能带在布里渊区中心点 简并。这样的材料容易通过应变从普通绝缘体转变到拓扑绝缘体。很多候选的 Heusler 化合物(LnAuPb、LnPdBi、LnPtSb、LnPtBi)包含稀土元素Ln,其有强关联 f 电子包含自旋序、轨道序等,导致磁性、超导电性和重费米子行为,使得实现诸多新的拓扑效应和新奇粒子成为可能。通常,XYZ 可以被视为有一个 Xn+ 填充闪锌 YZn-子格,其中价带电子数量与 YZn-相关联的等于 18(图 1-9)。18 电子化合物是闭壳的种类,没有磁性且具有半导体性(图 1-10)。
基于 Heusler 化合物的拓扑绝缘体自然具有 f 壳层稀土元素。在化学功能
(传递 3 个电子到闪锌晶格并决定晶格大小)外,未闭合的 f-壳层电子呈现出丰富的物性。例如,① LnPtBi 中发现的体态可以实现动力学轴子,其自旋波激发与电磁场相拓扑耦合,这样的效应提供了可调光学调制器新的设计;
② YbPtBi 中的重费米子行为可能实现最近提出的拓扑Kondo 绝缘体[64];③非中心对称低载流子 LaPtBi 系统中的超导性,这里空间反演对称的缺失理论上支持拓扑超导 [65]。
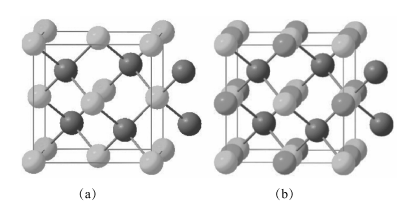
图 1-9 (a)闪锌矿结构;(b)填充闪锌结构后的 XYZ
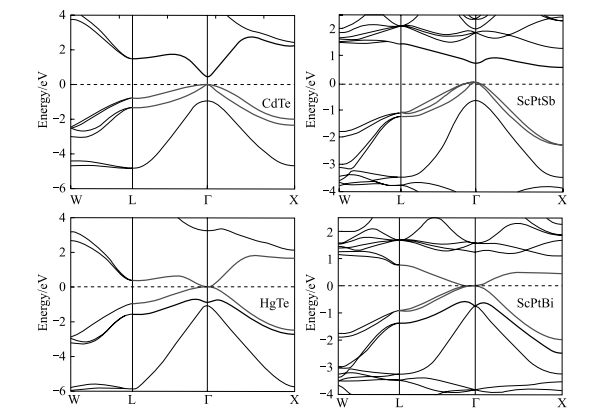
图 1-10 CdTe 和 HgTe 与 ScPtSb 和 ScPtBi 对比。红色表示 8 对称性, 蓝色表示 6(书末附彩图)
由于以上的电子拓扑态是由能带的拓扑不变量来决定的,因此光子晶格中冷原子气体和光子晶体中的能带也可以具有类似的效应。最近有人提出了尝试在超冷原子系统中观察简单量子霍尔行为,提出了利用一维 Harper 方程进行 Hofstadter 蝴蝶能谱的模拟 [66]。人们还设想利用类似的方案来实现单向传输的光子边缘态及其拓扑性质来实现新型功能光子器件。Hafezi 等 [67] 模拟了通过线性光学元素,利用二维的耦合共振光波导(CROW)网络构造量子自旋哈密顿量,发现量子霍尔系统的关键特征,包括特有的 Hofstadter 蝴蝶(Hofstadter butterry)图形和鲁棒边缘态。特别是他们证实了拓扑保护可以用来改进光延迟线的性能,以克服光子技术中一些和无序有关的限制。
他们提出用合适的光器件构成的CROW 实现拓扑保护的光子器件。他们模拟了一个二维磁紧束缚哈密顿量,考虑顺时针周转(赝自旋向下)和逆时针周转(赝自旋向上)两种模式作为赝自旋的两个分量。这不需要显见的时间反演对称性破坏。通过在二维系统排列中耦合这些模式,他们发现在合适条件下,这样一个光子系统可由采用紧束缚模型正方晶格的带电玻色子哈密顿量描述,但是增加了一项正交的赝自旋依赖的有效磁场:
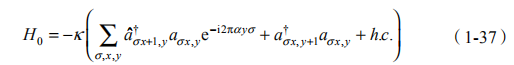
其中, 表示光学模式耦合概率。特别地, 光子绕晶片转一圈需要相位2 — 与含 个磁通量子等价。当顺时针和逆时针模式退简并时,就可以选择性地驱使每种模式,并在不破坏时间反演对称性的情况下,观测到边缘态行为。通过与电子整数量子霍尔效应进行直接类比,发现光子在系统的边缘态中传输,且对无序效应不敏感,因此可以构成鲁棒的光子器件。
在每个竖直的连接波导和谐振腔中考虑一对散射子(图 1-11),其对应的哈密顿量为
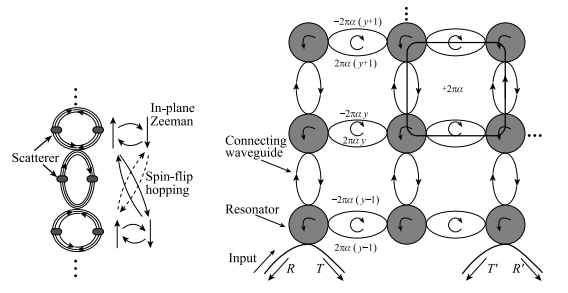
图 1-11 有合成磁场的光子系统模型
光子组成的量子霍尔系统中边缘态局域在两个 Hofstadter 带之间
(图 1-12)。在延迟线中,他们发现增加系统的周长,CROW 内的输运降低而边缘态的输运不受影响。
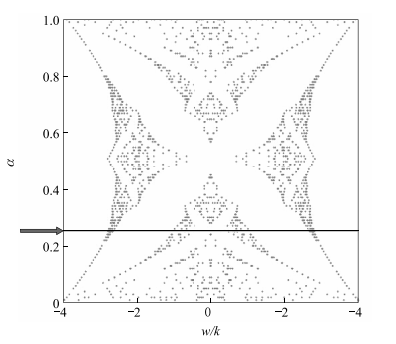
图 1-12 Hofstadter 蝴蝶能谱
在光子系统中实现磁性的许多设想中都要求有外场,比如大的外磁 [68]、应力 [69]、简谐调制 [70] 或者光力学诱导的非互易性 [71] 等。然而,已经发现这样的外场不是必要的。通过偏振方法 [72],差分光学路径 [67] 或是双各向异性超材料(metamaterial)[73],我们可以得到一个类磁哈密顿量,可与电子系统中的自旋轨道相互作用直接类比,实现光的拓扑态 [74]。特别是这样的系统在硅光子学中有直接的应用 [67,69]。利用诱导的赝自旋-轨道相互作用可以实现衍生的规范势。由于谐振腔的尺寸较大(几十微米),允许人们通过光成像对波函数进行直接测量。最近的实验测量(图 1-13)[75] 发现,①光沿体系的边缘态传播;②边缘态光传播的性质在宽的带中仍然保持,即对内禀无序不敏感;③边缘传播对外在的无序也是鲁棒的,实际上甚至在边缘谐振腔消失的情况下边缘态输运也不被阻止。
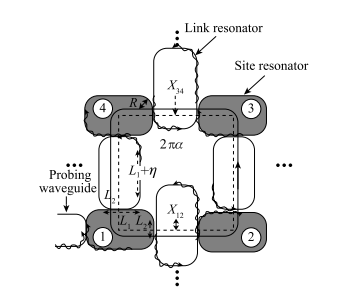
图 1-13 实验装置:在 34 和 12 之间区域,连接谐振腔垂直转变导致光子获得非零相位。实线表示光子沿逆(或顺)时针输运
实验中描述光子在晶片中跃迁的总体哈密顿量为
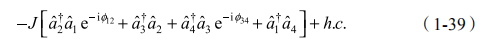
公式中,J 为跃迁概率;光子沿晶片逆时针运动获得相位 2 ,其中 =2n(x34-x12)/ 。如果晶片的相位在一个区域内是一致的,光子运动与带电粒子在均匀正交磁场下的运动相等价。可预测这样一个系统含有存在于区域边界的边缘态,如图 1-14 所示。在光子系统中,这样的边缘态可以通过驱使系统位于特定频率的带中来激发。这一平台可能为我们打开光子体系中由谐振腔引致不同类型的磁场和拓扑序方面的研究 [76]。结合强的非线性,人们进一步模拟和研究多体物理的效应。

图 1-14 均匀磁场下的边缘态输运
第五节 半导体异质界面能带调控引发的新奇量子相变
2000 年诺贝尔物理学奖得主赫伯特 克勒默(Herbert Kroemer)就预言“界面就是器件”,如今已成为全世界半导体科学技术领域内经常使用的一句口头禅。作为硅集成电路基本单元,初期的场效应晶体管,无论是金属-二氧化硅-半导体(MOS)器件,还是互补式的金属-二氧化硅-半导体
(CMOS)遇到的最大挑战是:如何控制 SiO2/Si 界面处的界面态数量及其稳定性,它决定了能否用 CMOS 器件构建成大规模硅集成电路芯片。这是 20 世纪 70 年代初“界面就是器件”的确切含义——控制 CMOS 器件中 SiO2/Si 界面态的数量和稳定性。将近半个世纪过去了,正因为人们能很好地控制了SiO2/Si 界面的性能,今天才有了过去无法想象的超大规模集成芯片。
近年来,“界面就是器件”这句话增添了全新的内涵。人们发现利用分子束外延生长(MBE)或金属有机物化学气相沉积(MOCVD)技术,可以制备高质量的极性界面。利用不同半导体材料界面间的电荷转移、应变、轨道杂化和再构以及晶格对称性的差异等因素已经可以从量子层面进行界面设计,能在界面处产生极强的局域极化电场和形成高浓度的二维电子气和/ 或空穴气。更重要的是,能在 1eV 的范围内显著地改变和调控半导体的能隙, 甚至可以将一些主流半导体材料变成半金属、拓扑绝缘体和拓扑半金属,以及激子绝缘体等形形色色的量子相。因此,这些极性界面为半导体物理提供了全新的实验平台,其奇特的物性也极有可能用来构造出全新的半导体电子和光电子器件。
追溯制备极性异质结的历史,1982 年,日本电工实验室 Yoshida 等尝试在蓝宝石衬底上生长制备 GaN-AlN 极性异质结 [77]。由于 GaN 与 AlN 晶格相对较为匹配,因此当 GaN 生长在外延 AlN 薄膜上时晶体质量较高。Yoshida 等利用蓝宝石(0001)面做基底,1200 下将单晶 AlN 薄膜用 Al 蒸汽分子束生长在基底表面,随后冷却到 700 ,并利用 Ga 分子束将 GaN 长在AlN 薄膜上。这种通过 MBE 方法生长得到的 GaN 薄膜的阴极发光强度比用GaN-蓝宝石异质结的高几十倍。同时,它的高 Hall 电导表明这些薄膜具有较好的晶体质量,如图 1-15 所示。
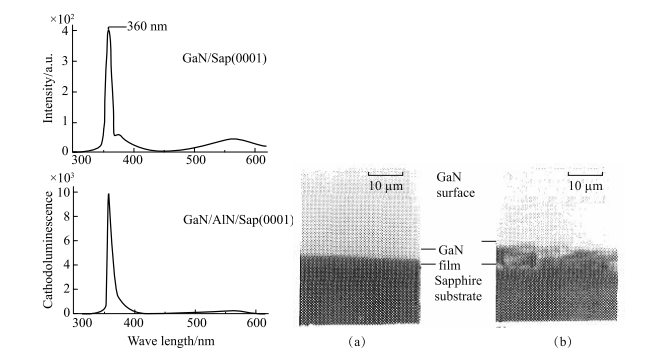
图 1-15 利用缓冲层生长的 GaN 薄膜与直接生长在蓝宝石
1985 年,日本川崎实验室Amano 等 [78] 在前面工作的基础上进一步改进了生长方法,采用低压MOVPE 方法在 AlN 缓冲层上成功地生长出表面平整且没有裂纹的单晶 GaN 薄膜。这种利用缓冲层异质结制备 GaN 薄膜的方法成功改进了GaN 薄膜的晶体质量与电学特性, 奠定了 GaN 基光电器件的基础。1991 年美国阿帕光学公司Khan 等[79] 利用 MOCVD 方法得到了 GaN/ AlGaN 异质结。与单纯的 GaN 薄膜相比,其电子迁移率提高了几十倍(图 1-16)。多层异质结在 77K 下迁移率可高达 1980cm2/(V s)。但是,人们并没有去测量界面处的局域极化电场,弄清迁移率提高的原因。
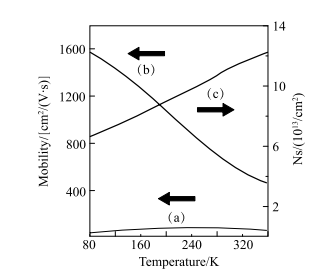
图 1-16 3000 GaN 层载流子迁移率(a)、
3000AlGaN/GaN 层载流子迁移率(b)、
3000AlGaN/GaN 异质结载流子面密度(c) 随温度变化关系
近年来界面领域内一个新兴的研究方向是过渡金属氧化物的界面,随着氧化物薄膜生长技术的发展,科学家已经能有效控制过渡金属氧化物的界面。在这样的氧化物界面上,过渡金属的 s 电子转移到氧离子上,界面的物理性质主要由关联性更强的 d 电子决定,使得过渡金属氧化物的界面表现出完全不同于其体材料的新奇特性。比如 2004 年,美国贝尔实验室 Ohtomo 等在单晶(001)SrTiO3 衬底上长出了 LaAlO3/SrTiO3 异质结,并通过精确控制界面处的原子种类分别实现了空穴掺杂和电子掺杂。在空穴掺杂情况下,界面呈绝缘性;而在电子掺杂情况下,界面则呈导电性且具有很高的载流子迁移率,并出现磁阻振荡现象 [80]。后续的系统研究更揭示出,通过调控界面对称性以及 d 电子的自由度,可以在过渡金属氧化物界面产生丰富的层展现象。比如,破坏界面的空间反演对称性将导致绝缘体-金属转变 [80];破坏界面的时间反演对称性会引致磁性 [81];破坏规范对称性则会导致超导相出现 [82]。2012 年,Nature Materials 刊发氧化物界面专辑,详细阐述了这种层展现象 [83],并认为“界面仍旧是器件”(图 1-17)。
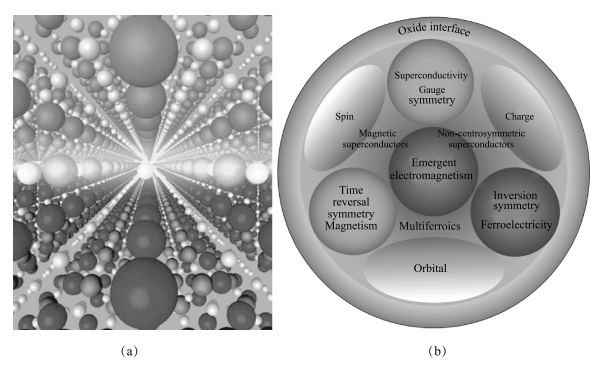
图 1-17 (a)绝缘体 LaAlO3/SrTiO3 异质结界面上出现高迁移率的导电层;(b)调控氧化物界面对称性与关联电子自由度可以产生丰富的层展现象
以前的研究表明,利用半导体材料原子的电负性不同,沿极性面方向生长材料,可以实现高浓度的二维电子气。例如,2007 年日本筑波大学研究组成功制备了含有单层 InN 的 InN/GaN 多量子阱,并在室温下观察到波长为350nm 左右的荧光峰(图 1-18)[84]。这些实验发现均表明,氧化物极性界面会呈现多种多样的新奇效应,但是对它们的物理机制并不清楚。
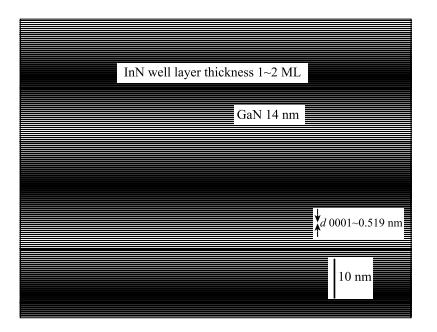
图 1-18 InN/GaN 多量子阱 XTEM 图像
2012 年,中国科学院半导体研究所超晶格国家重点实验室常凯理论小组与美国加州大学圣塔芭芭拉分校的苗茂生合作,提出了在 InN/GaN 量子阱中可以产生极强的局域电场,可达 10 MV/cm 量级。该电场可在 0~2 eV 大范围内调控半导体材料的带隙 [85]。InN 和 GaN 虽然同为 - 族氮化物,具有相同的空间点群,但二者的晶格常数却相差较大。界面应力所带来的压电效应会在垂直于界面的方向上形成巨大的内建极化电场。通过第一性原理计算的预测,该电场高达 10 MV/cm。并且,通过改变 InN 层厚可以调节电场,预计可在 0.8 eV 到 0 eV 的大范围调控带隙,甚至导致能带翻转,实现拓扑绝缘体相(图 1-19)[85]。2013 年美国 AMES 国家实验室和亚利桑那州立大学实验组发现,InN/GaN 量子阱的极化场会导致光致发光峰随 GaN 势垒层厚度而偏移(图 1-20)。这是因为随着 GaN 势垒层厚度增加,势垒层内部的极化电场有显著的降低。这样可以使 InN 发光设备在室温下覆盖有用波段的光 [86]。该实验证实了理论课题组关于内建电场强度的计算和预测 [85]。2014 年,美国桑迪亚国家实验室的潘伟和诺贝尔奖得主崔琦研究组的实验声称,在 InN 单层和 10nm GaN 势垒层组成的 40 个量子阱超晶格结构中得到了不依赖于温度的、具有较高迁移率的二维电子气,如图 1-21 所示, 他们认为观察到了拓扑转变的迹象 [87],在他们的文章中多次大段引用理论工作 [85] 来解释和理解实验结果。
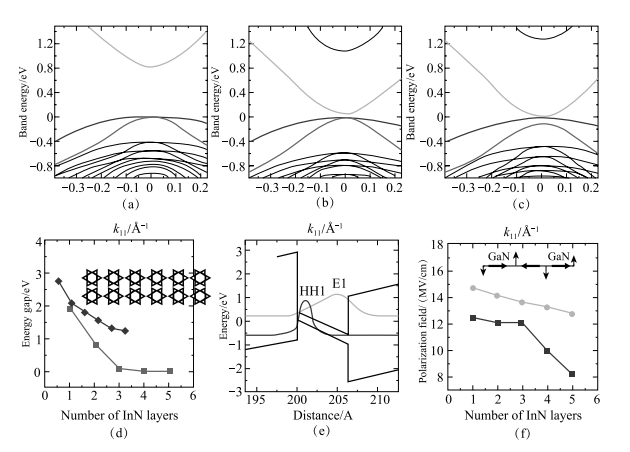
图 1-19 GaN/InN/GaN 量子阱系统带隙与 InN 层厚关系,改变 InN 层厚,可以实现近1eV 调节
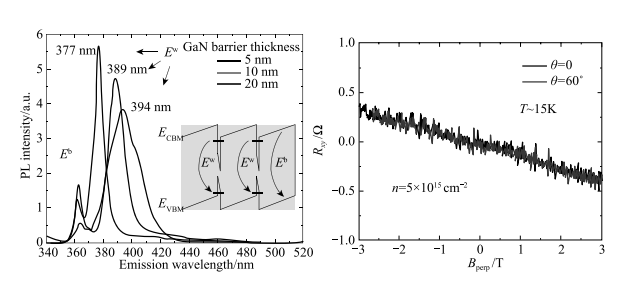
图 1-20 GaN 势垒层对荧光峰波长的调制 图 1-21 表示平面法向和所加磁场的夹角,从图中可以看出不同角度的霍尔电阻相重合,体现出电子气的二维性质常凯理论小组又研究了晶格匹配的主流半导体材料(如锗、砷化镓)之间的界面。他建议通过外延生长的方式,沿[111]极化方向生长 GaAs/Ge/ GaAs 量子阱 [88]。一方面,通过布里渊区折叠可使锗由间接半导体变为直接半导体;另一方面,通过确保一侧的界面是 As—Ge 原子成键,而另一侧为Ga—Ge 原子成键,就会在超薄的 Ge 原子夹层区域上产生一个强大的自建电场(图 1-22)。
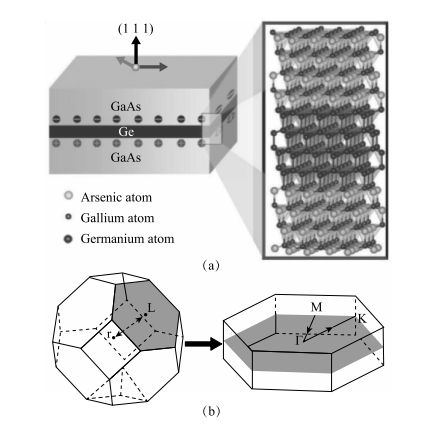
图 1-22 (a) 砷化镓夹层超薄锗原子层的人工微结构。其中,右上方的放大图例显示了夹层为四双原子层锗的砷化镓 / 锗 / 砷化镓量子阱的原子构型。尤其需要注意的是,在锗原子层的两侧分别对应镓原子层和砷原子层。正是这种结构的不对称引起了如左图所示的电荷累积。(b) 沿[111]晶向生长的砷化镓 / 锗 / 砷化镓量子阱的布里渊区示意图,从此图中我们可以看到闪锌矿化合物布里渊区的折叠情况
这个电场来源于 As 原子和 Ga 原子的价电子数差异,As 有 5 个价电子, 而 Ga 只有 3 个价电子,这样,在 As-Ge 界面将积累电子,在 Ga-Ge 界面会累积空穴,相当于在锗原子层产生很强的局域内建电场。该内建电场导致锗原子层能隙减小。当内建电场足够强时,可以期待系统的能隙变成负带隙, 并且产生很强的自旋轨道耦合,从而能打开较大的非平庸带隙,并进入拓扑绝缘体相。
他们第一性原理计算的结果表明,在 GaAs/Ge/GaAs 量子阱中,由于量子限域效应和内建电场的存在,内建电场可以高达 14 MV/cm。并且,通过改变层厚,系统能隙可以由 1.6 eV 变化至 0 eV,即从可见光到 THz 的范围变化。值得注意的是,在该量子阱系统中,应力可以作为辅助手段使能隙连续变化,这样的方案可用来制作宽谱光电器件(图 1-23)。
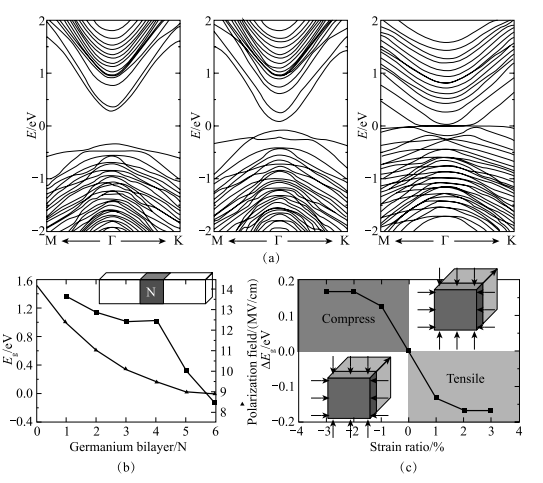
图 1-23 (a)不同锗组分的 GaAs/Ge/GaAs 量子阱杂化密度泛函能带,从左到右分别为2 个锗双原子层,4 个锗双原子层以及 3% 平面内张应力下 4 个锗双原子层结果;(b)锗双原子层层数与带隙(三角形线)及内建极化电场强度(方形线)的关系;(c)应力对带隙的调控
应当强调,这类极性界面调控方案,不但可以在大范围内调控主流半导体的能隙,而且具有普适性和重要的应用前景。近来,常凯小组又证明利用界面能带调控还有可能实现拓扑半金属相、自旋半金属相 [89] 等多种新奇量子相。近年来凝聚态物理的发展揭示了许多新奇的物相,如拓扑绝缘体(topo- logical insulator)、拓扑半金属(topological semimetal)以及自旋半金属(half- metal)等。由界面调控方案产生的极强内建电场,能够在 1 eV 的量级上大范围调控能隙,正好提供了引发主流半导体发生新奇相变的关键手段 [90,91],使主流半导体材料中可以展现一些原本在重元素或含有 d-或 f-电子的材料体系中才有的量子相。
极性界面的量子调控方案已经开始成为半导体物理重要的前沿领域。密度泛函理论先驱、美国科罗拉多大学教授 Alex Zunger 等已将这一方案应用于 - 族和 - 族化合物半导体异质结 [92] 及二维材料黑磷 [93] 中,均实现了上述材料的拓扑绝缘体转变。最近,Review of Modern Physics 关于拓扑能带理论的文章 [94] 已经将通过极性界面设计作为开辟 2D 材料拓扑相的第四类新方案。同时,将 GaAs/Ge/GaAs 系统单独列为有希望的拓扑材料重点介绍给读者。第一类是 Kane 和 Mele 提出的在石墨烯上吸附重原子;第二类是光子晶体中光子的拓扑态;第三类是叠加符号相反 Rashaba 自旋轨道耦合的二维电子气。《物理学进展》最近在关于二维材料拓扑相的综述文章中推介了极性界面方案 [95]。
总而言之,随着半导体器件尺度的不断缩小,界面效应愈发明显。已有的研究成果表明,界面调控可以在很大范围(0~2 eV)内调控主流半导体材料的带隙。这一方面将会大大拓宽主流半导体材料的应用范围;另一方面, 由于极性界面所提供的 10MV/cm 量级的内建电场是界面生长的自洽结果, 多种诸如自旋半金属相、拓扑绝缘体相、拓扑半金属相等新奇相变的实现成为可能。这方面的工作是由中国科学家引领的,为界面能带调控、新奇量子相变调控等基础物理研究提供了全新的方向和领域,应当给予大力支持,以期从实验上获得重大突破。
第六节 展望
半导体能带计算方法的发展目标在于解释和预测半导体材料及其微纳结构的电子结构,发展普适的第一性或半经验的计算方法与理论,不断改进方法以精确地计入库仑相互作用、范德瓦耳斯相互作用等多种因素对电子结构的影响。
半导体能带第一性原理计算方法历经近半个世纪的发展与沿革,在陆续考虑 GW 近似或杂化泛函 HSE 的修正后,已经可以准确地预测绝大多数半导体材料力学和电子结构性质。第一性原理的计算方法虽然较精确,但是仍然受到计算量的限制,难以计算原子数目巨大的半导体纳米结构的电子结构。近年来人们开始发展无轨道的密度泛函理论(orbital-free density functional
theory)来计算包含百万电子体系的能谱,这套理论基于Thomas-Fermi 模型, 通过直接计算电子密度为基础预测体系能带结构,其计算精度高度依赖于动能的密度泛函,近年来利用蒙特卡罗取样 [96]、半局域近似 [97] 等手段在动能密度泛函的精确性与可拓展性方面获得了长足的进步,是半导体电子结构计算方法值得期待的发展方向之一。
半导体能带计算的方法发展开始与计算硬件平台相结合,众核计算平台,尤其是目前新兴的 GPU 加速科学计算在半导体电子结构预测方面极为成功。究其原因在于:① GPU 的双精度浮点运算能力较传统的 CPU 计算核心显著增强,为更高精度要求的第一性原理计算提供了基础条件;② GPU 的带宽更宽,满足第一性原理计算中常用的快速傅里叶变换(fast Fourier transform,FFT)等带宽密集型计算;③ GPU 由于其在图形渲染等方面广泛的应用,催生出了众多的数值计算库(如 CUBLAS、MAGMA、CUDPP、CUFFT 等)的不断发展,为使用 GPU 完成 PWP-DFT 计算提供了便利条件。现在,与 GPU 结合的第一性原理计算方法正经历从特殊定制的机器语言向更为易用的高级语言(如 CUDA、OPENCL 等)发展,这些语言将会方便地被C、C++、Fortran 等传统高级语言调用,极大地增强现有半导体电子结构计算方法的计算能力。
另外值得注意的新动向是,人们开始采用遗传算法以及基于人工智能的机器深度学习方法预测材料晶体结构和电子结构 [98-100]。这些新的计算方式将极大地降低计算能力要求(使计算需求由N3 增长依赖关系降低为线性增长),显著提升半导体预测效率,并革命性地提高和拓展新型半导体材料结构的搜寻方式。可以期待的是,半导体能带计算方法必将伴随半导体物理的深入研究保持着旺盛的生命力。
最后,希望重点推介的是利用不同半导体材料界面间的电荷转移、应变、轨道杂化和再构,以及晶格对称性的差异等因素已经可以从量子层面进行界面设计,能在界面处产生极强的局域极化电场和形成高浓度的二维电子气和 / 或空穴气。更重要的是,这类极性异质界面能在 1 eV 的范围内显著地改变和调控半导体的能隙,甚至可以将一些主流半导体材料变成半金属、拓扑绝缘体和拓扑半金属,以及激子绝缘体等量子相,成为研究新奇量子相变的新体系。各种奇特量子相的物性也极可能用来构造出全新的半导体电子和光电子器件。
常凯(中国科学院半导体研究所,中国科学院半导体超晶格国家重点实验室)